
Bioinformatics pipelines: The arteries and veins of modern NGS data analysis
The challenges come with scale and formats of genomics data, inefficient and non-replicable experiments and their results, handling intermediate data etc.

Introduction
Pipelines are transportation systems used to move goods and materials such as sewage, water, and energy like natural gas, biofuels, and liquid petroleum1.

In software engineering, a pipeline consists of a chain of processing elements (processes, threads, coroutines, functions, etc.), arranged so that the output of each element is the input of the next2;

Bioinformatics, specifically in the context of genomics and molecular pathology, uses computational, mathematical, and statistical tools to collect, organize, and analyze large and complex genetic sequencing data and related biological data. A set of bioinformatics algorithms, when executed in a predefined sequence to process NGS data, is collectively referred to as a bioinformatics pipeline3.

RNA seq pipeline with bioinformatics tools on left and functionality on right4
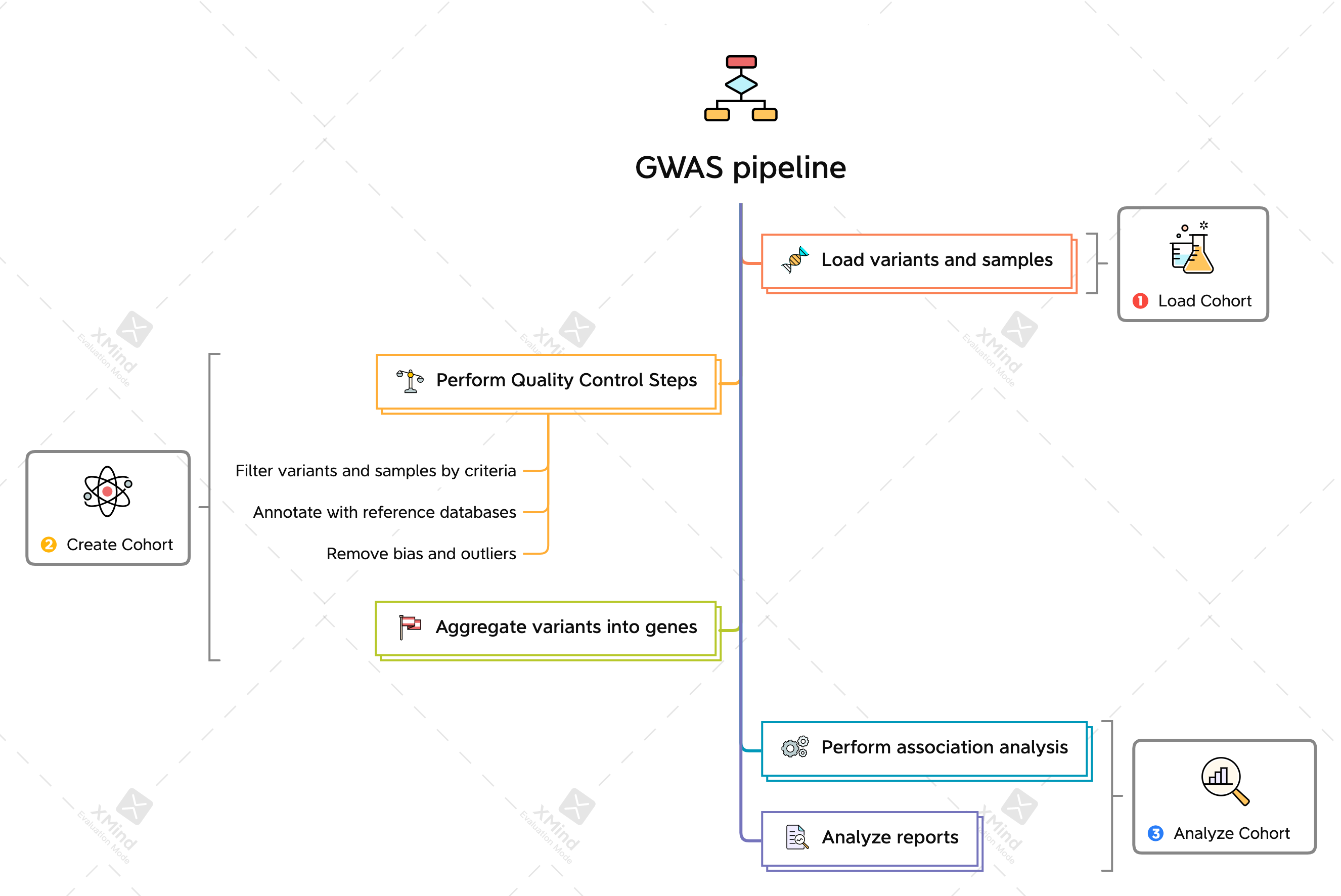
Genome-Wide Association Studies(GWAS)
Association study between variants of genes(alleles) and phenotypes
Phenotype ~= Covariates + Genotype
Correlation analysis between SNPs(IDs like AA, AT, TT) - alleles and phenotype(like BMI)
identify genes involved in human diseases.
searches the genome for small variations, called single nucleotide polymorphisms or SNPs (pronounced “snips”), that occur more frequently in people with a particular disease than in people without the disease.
Why GWAS?
pinpoint genes or markers that may contribute to a person’s risk of developing a certain disease.
understanding variations that affect a person’s response to certain drugs
understanding interactions between a person’s genes and the environment.
Laying the groundwork for the era of personalized medicine.
Identification of novel SNV-trait associations
highlighting the molecular pathways underlying disease, providing potential targets for therapy
Computational Challenges of GWAS
With ever-increasing data generated by Next Generation Sequencing(NGS) pipelines, it has become difficult to store and process data on an on-premise cluster(which further needs to be maintained). The cyber attacks on biotech firms have further fueled the debate to move data to cloud for storage and computing5 6.
Many biotech firms have realized the realities and are moving their data to cloud7.
To give you an estimate of UK BioBank(UKBB) dataset size in 2021
Structured Data(Text): 50GB
Imaging data: 450 TB
Unstructured Data: 50 PB
Whole Exome Sequencing(WES): 1.5 PB
Whole Genome Sequencing(WGS): 12.5 PB
Overall data: 15 PB(60 fold increase as expected in 2016)
Proteomics and Metabolomics: 50 PB by 2025
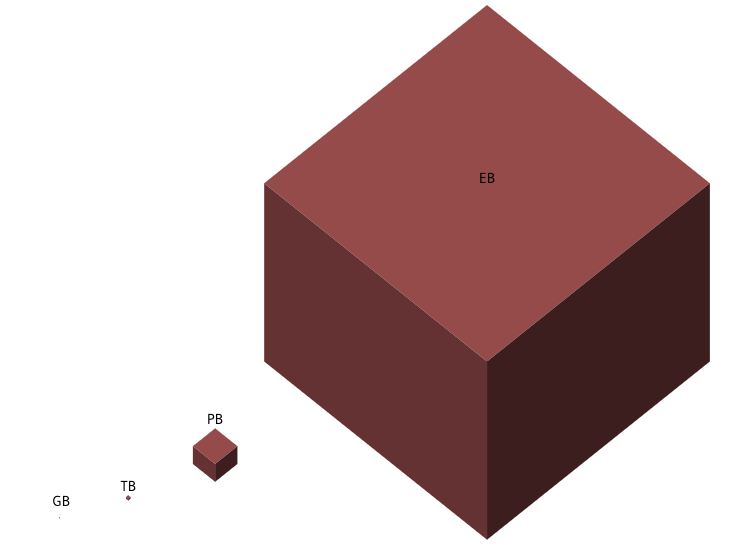
If you can see the GB, congrats, your monitor is very large or very clean8
The cost of computing NGS data through a bioinformatics pipeline can vary from 50-300 USD per sample or 17-170 USD per hour. The cost can rack up to millions for getting qualitative results9.
Costs can be brought down by
shifting data to glacier mode when not in use
deleting intermediate datasets after use(as they can be reproduced)
Building cached, modular, and reusable bioinformatics pipelines
UI interface to access and visualize data for interpreting downstream results
Kubernetes
Kubernetes is an open-source platform for automating deployments, scaling, and operations of application containers across clusters of hosts, providing a platform for container-centric infrastructure. Kubernetes ensures that container orchestration can scale properly, mask failures, and maximize system resources.
When scaling containers in a large-scale production environment, developers are usually tasked to develop container scheduling systems, monitoring of containers during run time, design auto-scaling and load balancing systems for the container instances, and determine if zero downtime is required for deployment.
Therefore, the automated coordination of these systems and processes is very important but could be very complicated. Kubernetes could help in achieving these goals.
How Kubernetes can help in bringing costs down for genomic pipelines like GWAS?
Most of the cloud cluster providers provide a fixed cluster. Some tasks of your genomic pipelines may require different computation power. Therefore, the nodes may stay idle getting billed without getting used.
Kubernetes can give flexibility of scaling up of nodes on demand as compared to predefined cluster optimising based on compute power required. Also using on-spot as compared on-demand instances can bring down the cost significantly10.
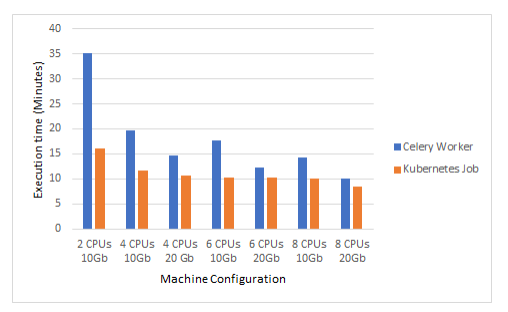
The scalability of a Kubernetes job is superior to the scalability of Celery11
Apache Spark on Kubernetes
Apache Spark is an open-source distributed computing framework. In a few lines of code (in Scala, Python, SQL, or R), data scientists or engineers define applications that can process large amounts of data with Spark taking care of parallelizing the work across a cluster of machines.

You can submit Spark apps using spark-submit or using the spark-operator12
Hail on Apache Spark
Hail is an open-source library that provides accessible interfaces for exploring genomic data, with a backend that automatically scales to take advantage of large compute clusters. Hail enables those without expertise in parallel computing to flexibly, efficiently, and interactively analyze large sequencing datasets.
Hail is the analytical engine behind projects such as the Genome Aggregation Database, the UK Biobank mega-GWAS, eQTLs in GTEx, TOPMed, the Psychiatric Genomics Consortium, and the Centers for Mendelian Genomics.
PS: This is a two part blog series. The challenges of deploying a GWAS pipeline on AWS cloud using Kubernetes will be shared in next blog.
References
GWAS: https://medlineplus.gov/genetics/understanding/genomicresearch/snp/
GWAS using hail: https://www.broadinstitute.org/videos/broade-hail-practical-2-genome-wide-association-studies-gwas-and-rare-variant-burden
Footnotes